Graph alignment uses all information about known variants
Genomic alignment is the process of matching read data from a DNA sequencer to a known reference genome.
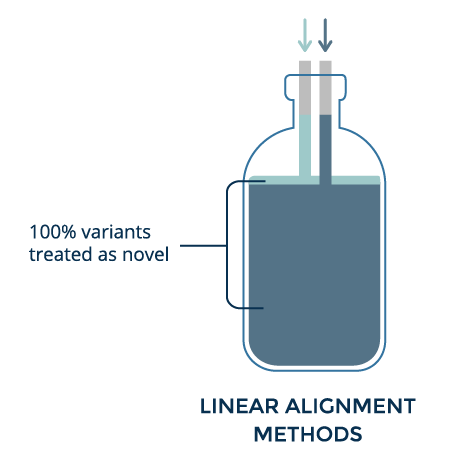
Linear alignment helps identify variants by highlighting places where aligned reads differ from the reference. However, when the alignment differs too much, that location is discarded. Thus, the more an individual differs from the reference, the less accurate variant calling becomes.
With graph alignment, which uses all the information about known variants, most reads can be matched — even those containing complex variants.

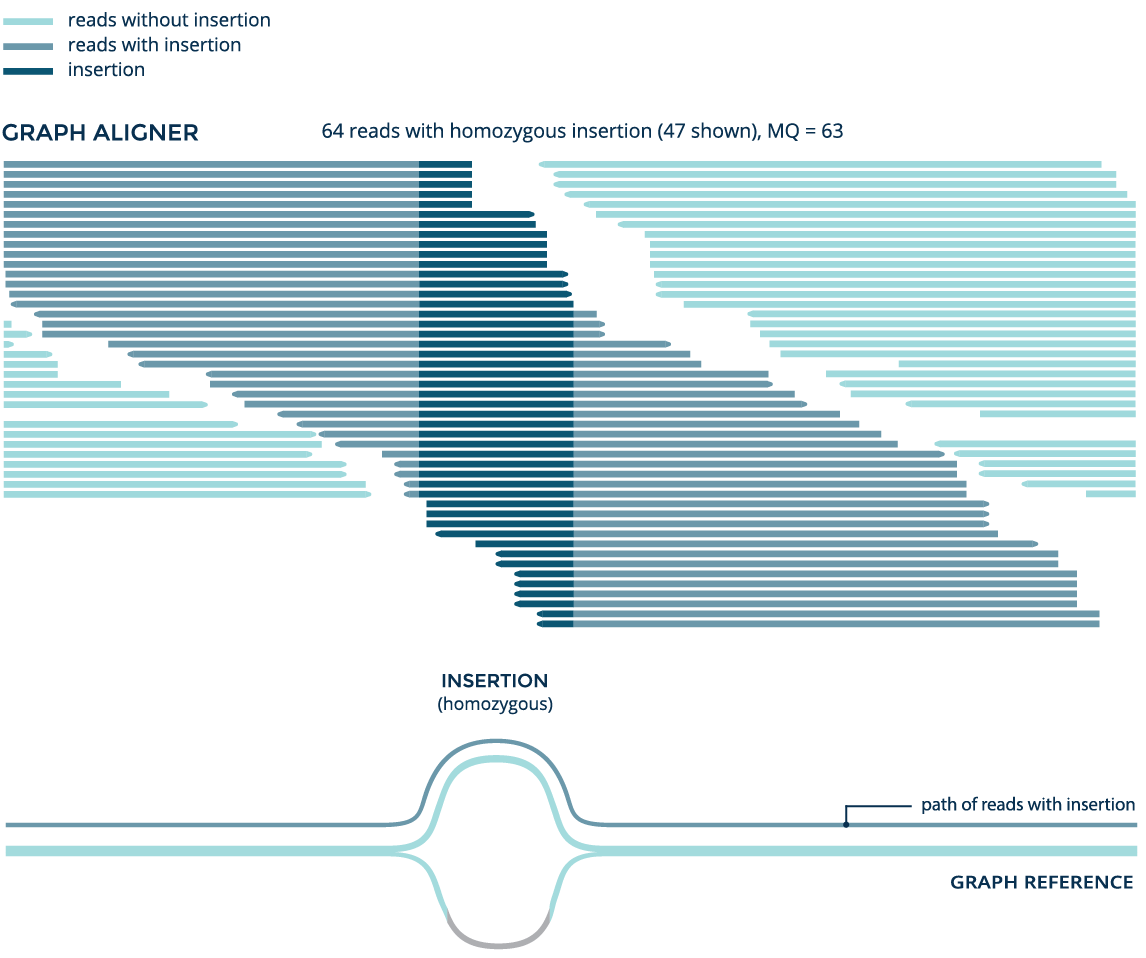
Graph improves alignment for a known variant
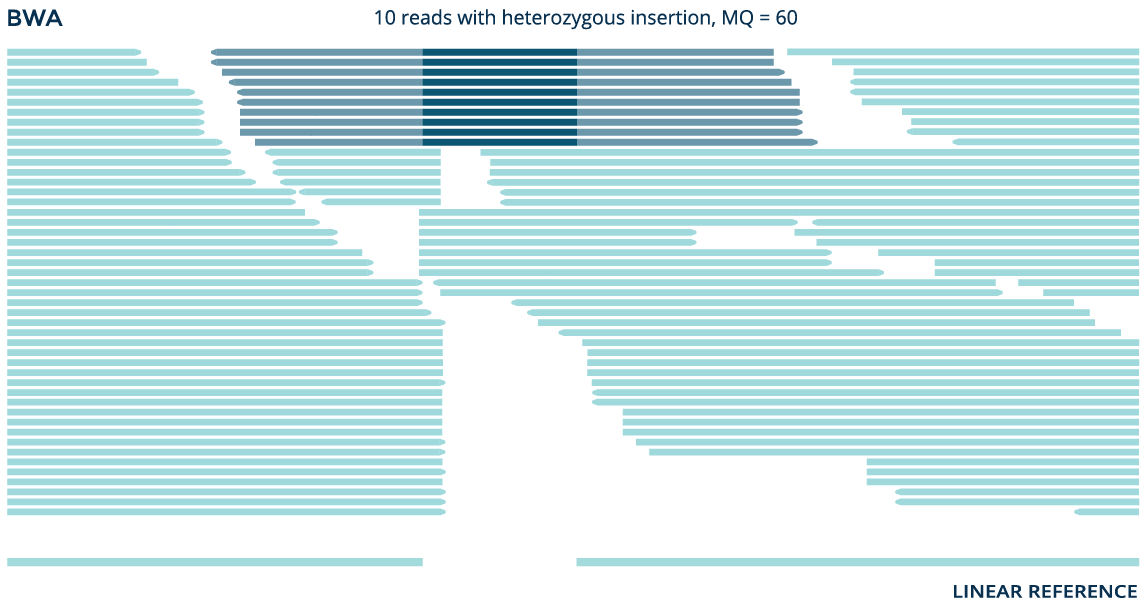
Using data from the whole genome sample NA12878, both the graph aligner and the linear aligner BWA identify a known 41bp insertion (rs141252781). But the graph aligner aligns significantly more reads and accurately detects the variant as a homozygous insertion, while BWA erroneously detects it as heterozygous.
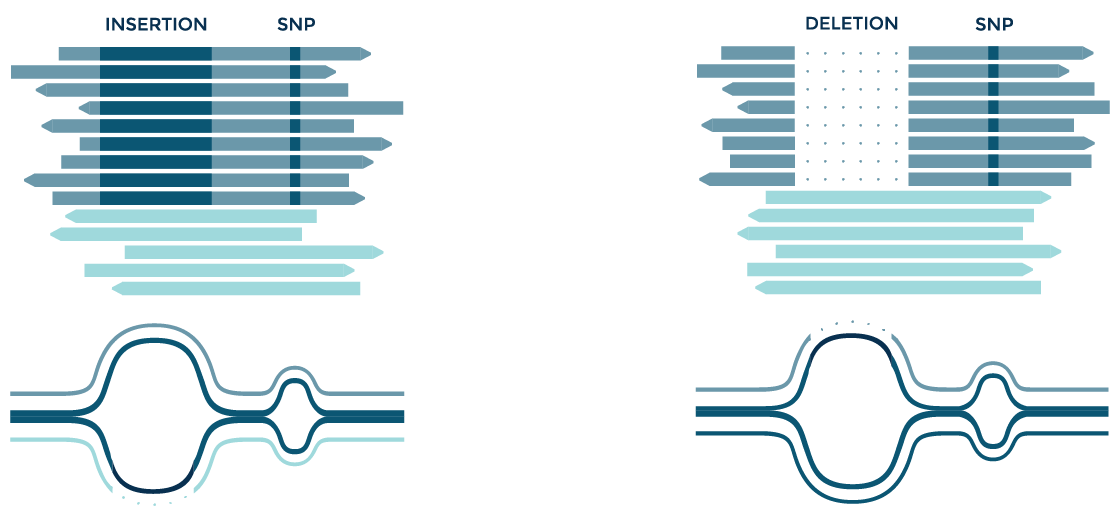
Identifying complex variants
SNPs often occur near larger variants such as insertions and deletions. SNPs are thus often missed in these regions when reads contain large mismatches. Graph alignment facilitates the discovery of in-phase variants, and other types of complex events.
Identifying variants within variants
In high variability regions of the genome, long alternative branches continue to evolve over time. A graph reference easily places variants within existing variants to facilitate alignment.

Reference
NA12878 DNA from LCL; https://catalog.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM12878