Sentieon Multi-instance Whole Genome Workflow
Organizations have adopted the use of Next Generation Sequencing (NGS) as a one of the primary tools of their discovery, diagnostic and clinical efforts. Simultaneously — the number of tools available for NGS analysis has ballooned, with each tool having different capabilities for speed, accuracy, cost, etc. Seven Bridges offers more than 350 workflows and tools for NGS analysis, but more importantly helps organizations choose an optimal analytical tool for their specific job to be done. Starting with the right tool on a powerful biomedical data analysis platform minimizes the time to results and maximizes the probability of success.
One common need for clinical use cases and organizations with vast amounts of data is the following “configuration” — high accuracy and short processing time with a tradeoff of higher cost. An excellent tool to meet these requirements is the Sentieon toolkit. Here, Seven Bridges has developed a configurable Whole Genome workflow using Sentieon tools with reduced runtime on the Seven Bridges Platform by introducing configurable number of parallel instances.
Conceptually, in the new workflow sequence reads are split into an optimal number of groups on the fly, and each group is processed independently on an individual instance (Figure 1). After processing, all BAM files are merged together for removing duplicates step and afterwards processed in parallel by splitting it to chromosomal regions.
The execution flows of the Single-Instance and Multi-Instance Sentieon Variant calling workflows are shown below:
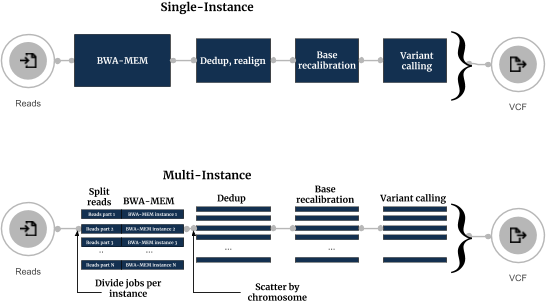
The most commonly used approach for running secondary DNA analysis on the cloud is done in several steps:
- Spawn a single instance from cloud provider.
- Transfer all necessary data to it: FASTQs, reference genome and other resource files, docker images from all of the tools and the workflow described in standardized syntax such as Common Workflow Language (CWL).
- Execute all steps in the workflow (alignment, deduplication, base recalibration, variant calling, variant filtering) with optimal usage of available processing, memory and storage resources by selected CWL Executor.
- Transfer all output data (BAM file, VCF, metrics) to a permanent storage space convenient for additional analysis.
Seven Bridges modified this approach by introducing the option to spawn multiple cloud instances on Seven Bridges platform and use them for processing one DNA sample. This multi-instance approach consists of the steps:
- Spawn multiple instance from cloud provider (e.g. 8).
- Transfer all necessary data to each of the instances (FASTQs, reference genome) and, for every instance, run the alignment and BAM sorting on different portion of paired FASTQ files.
- Receive all BAM files and perform removing of duplicates per chromosome.
- BAM files split by chromosome are passed to variant calling step after which obtained VCFs are filtered and merged.
- Transfer all output data (BAM file, VCF, metrics) to a permanent storage space convenient for additional analysis.
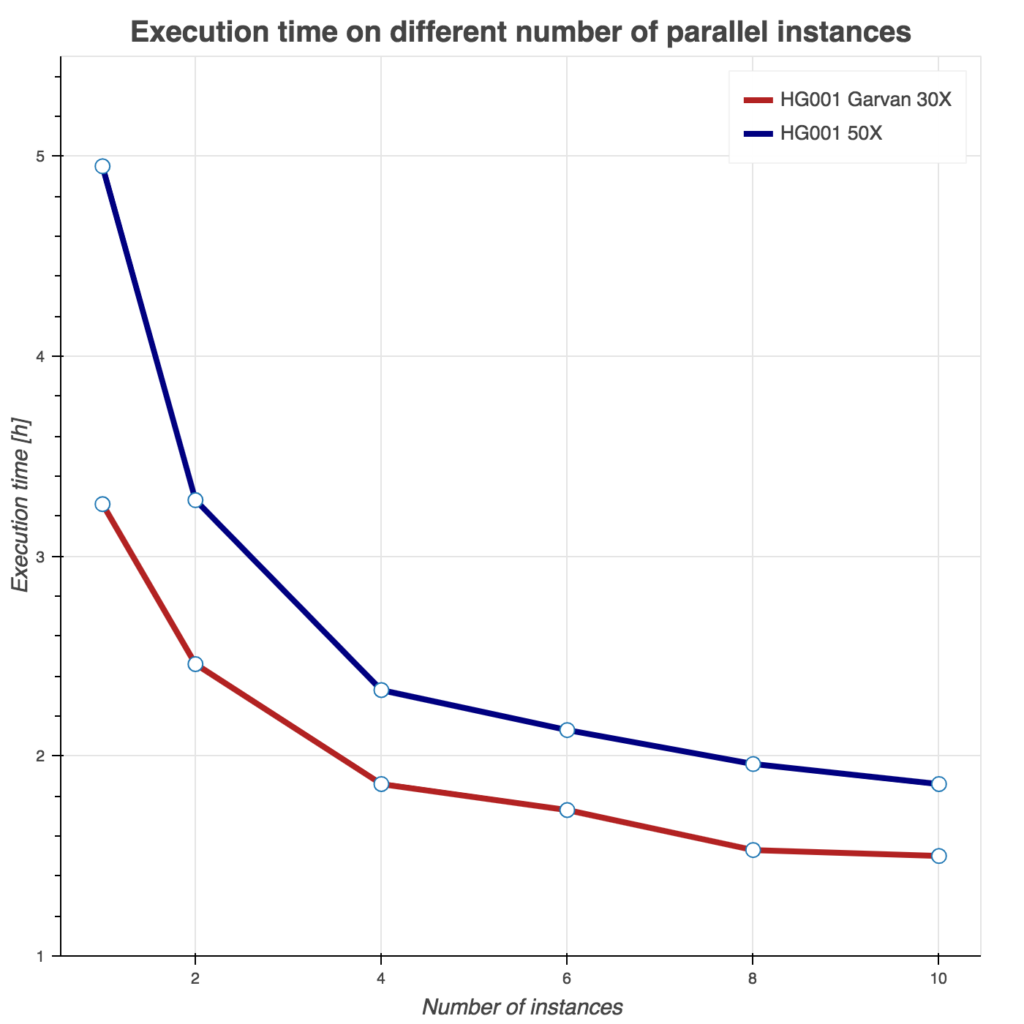
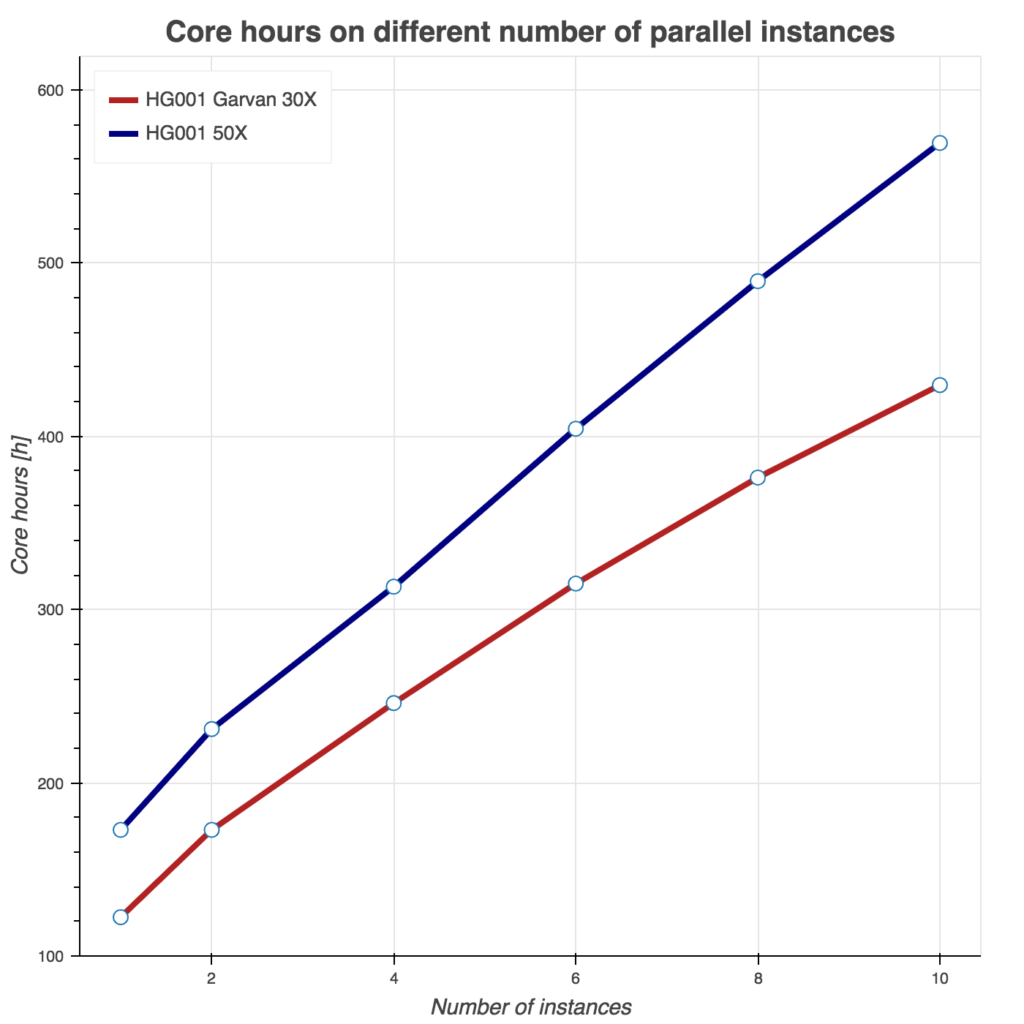
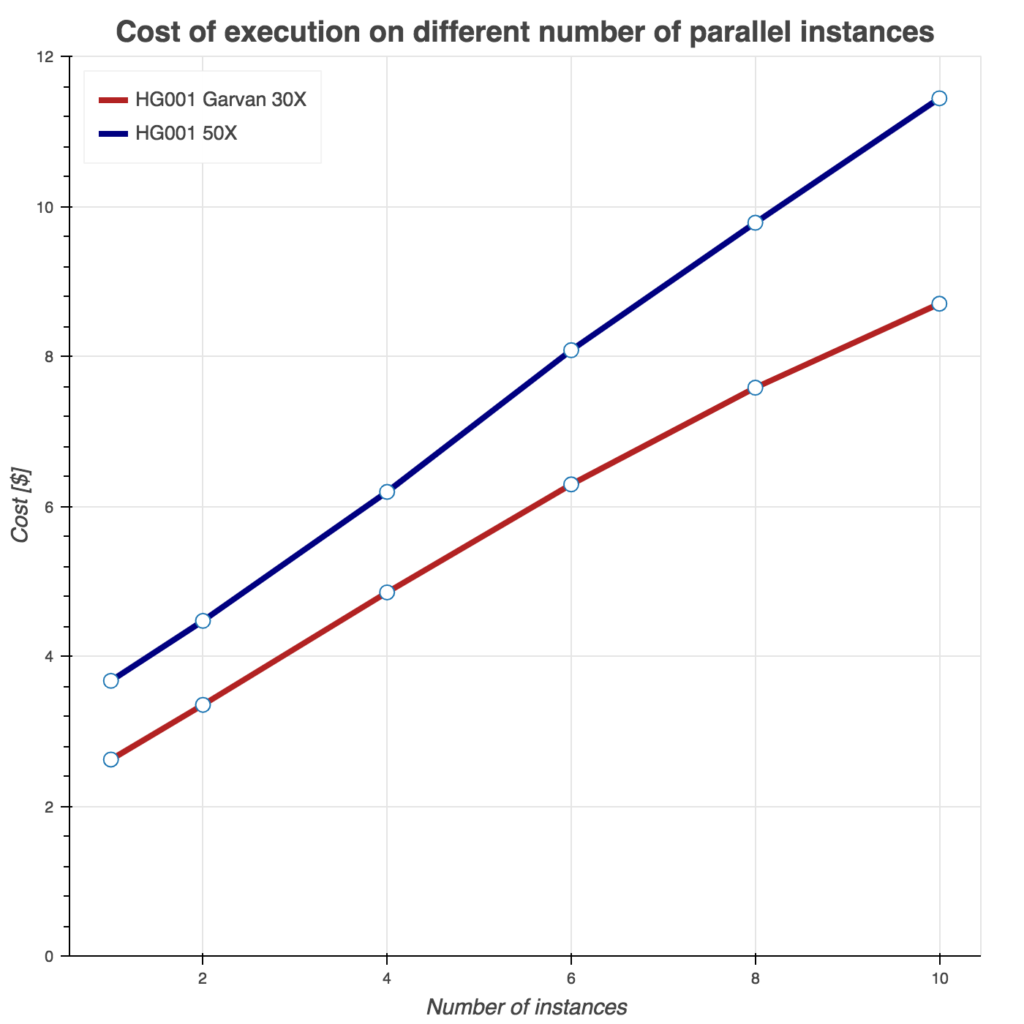
Several tests have been conducting to benchmark these two Sentieon workflows. First, we show runtime as a function of instances used in Figure 2. Similarly, we evaluate burned core (CPU) hours (Figure 3) and total compute cost (Figure 4) as a function of instances used. For all three tests, we used Amazon c5.9xlarge instance (36 cores, 72 GB of memory) and two samples from the FDA consistency challenge: 30x Garvan and PCR-free HG001 50x sample.
The improvements in execution time and cost are shown by benchmarking several 30x samples (sample names in Table 1) using eight c5.9xlarge instances. The results are shown in the Figure 5 and Table 1 below.
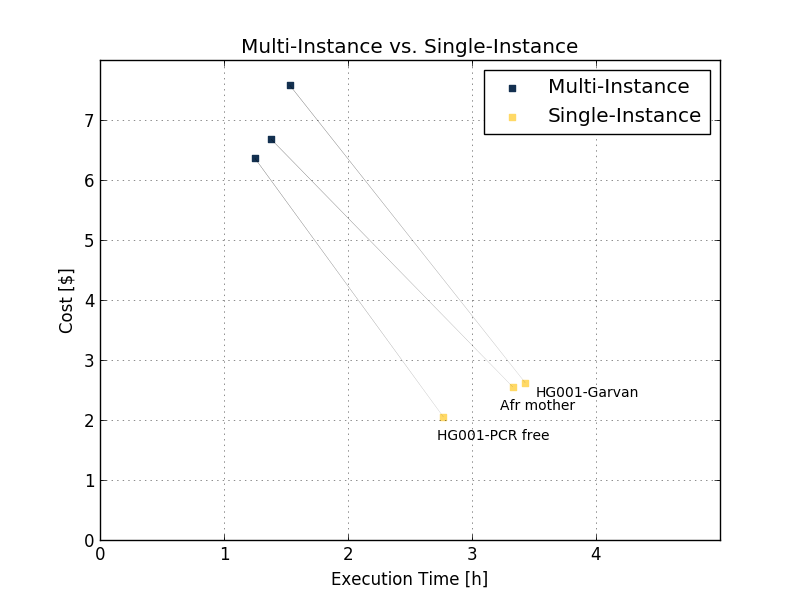
| African mother | HG001PCR free | HG001Garvan | ||
|---|---|---|---|---|
| Multi-Instance | Execution time | 1 h 23 min | 1 h 15 min | 1 h 32 min |
| Price ($) | 6.68 | 6.36 | 7.58 | |
| CPU hours | 333 | 301 | 376.2 | |
| Single-Instance | Execution time | 3 h 20 min | 2 h 32 min | 3 h 26 min |
| Price ($) | 2.55 | 1.94 | 2.62 | |
| CPU hours | 119 | 154 | 122.4 | |
The improvements in execution time and obtained cost are also shown on benchmarking of several 50x samples using eight parallel instances Execution time (Figure 6 and Table 2 below).
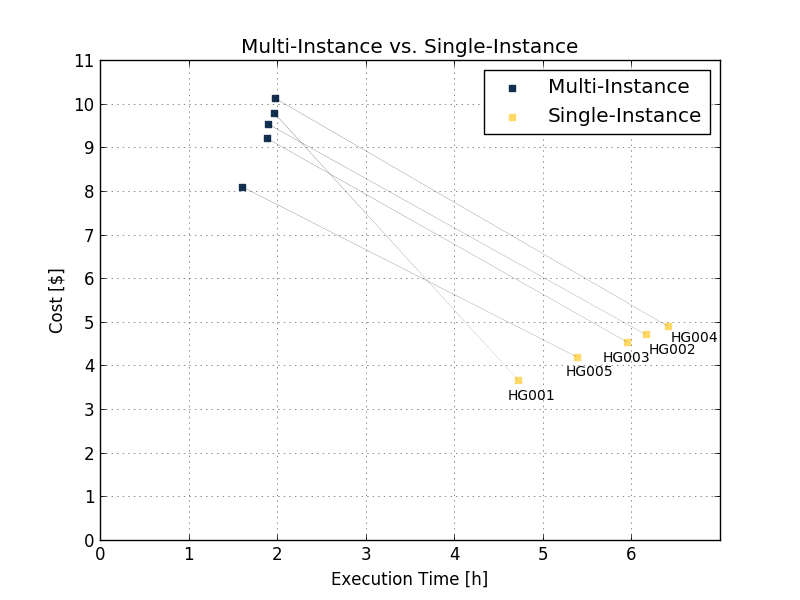
| HG001 | HG002 | HG003 | HG004 | HG005 | ||
|---|---|---|---|---|---|---|
| Multi-Instance | Execution time | 1h 58min | 1h 54min | 1h 54min | 1h 59min | 1h 36min |
| Price ($) | 9.78 | 9.53 | 9.21 | 10.12 | 8.1 | |
| CPU hours | 489.6 | 477 | 465.6 | 510 | 404.4 | |
| Single-Instance | Execution time | 4h 43min | 6h 10min | 5h 57min | 6h 25min | 5h 23min |
| Price ($) | 4.72 | 6.16 | 5.95 | 6.41 | 5.38 | |
| CPU hours | 262.8 | 192 | 283.2 | 311.4 | 262.2 | |
In order to validate variant call quality between the Single-Instance and Multi-Instance Sentieon workflows, we compared the results using Genome In A Bottle (HG001 and HG002) samples. The differences in precision, recall and f-score were less than 0.001%, which is expected due to stochastic effects (data not shown).
Another significant improvement in speed, from 40 minutes to only seven minutes for 30x sample, is using only the chromosome 20 interval to calculate the recalibration table, instead of the whole genome in Base Recalibration step. This optimization is done for both single and multi-instance workflows. From the tests we conducted on HG001 sample it decreases SNP f-score by 0.0069% (from 99.9176% to 99.9107%) and indel f-score by 0.0077 (from 99.2057% to 99.1980%), which is also in the margins of stochastic effects.
In summary, this new Sentieon Whole Genome workflow on the Seven Bridges Platform offers a configurable way for users to employ parallelization in order to achieve reduced run times for time-sensitive applications without losing accuracy or incurring substantial additional costs. Achieved average execution time for 30x samples is 1.38 h with average cost of $6.87, while for 50x the average execution time and cost are 1.9h and $9.35.
Interested in learning more about Sentieon tools and workflows or have questions about them? Feel free to contact us!